
ZOLGENSMA 2 x 10^13 génomes du vecteur-mL solution pour perfusion, boîte de 1 flacon (5,5mL ou 8,3mL)
Retiré du marché le : 27/05/2021
Dernière révision : 25/05/2020
Taux de TVA : 0%
Laboratoire exploitant : AVEXIS EU LIMITED
Source :

ZOLGENSMA est indiqué dans le traitement :
- des patients atteints d'amyotrophie spinale (SMA) 5q avec une mutation bi allélique du gène SMN1 et ayant un diagnostic clinique de SMA de type 1, ou
- des patients atteints de SMA 5q avec une mutation bi allélique du gène SMN1 et jusqu'à trois copies du gène SMN2.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Composition.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Immunité contre l'AAV9 préexistante
Un développement d'anticorps anti-AAV9 peut se produire après une exposition naturelle. Plusieurs études portant sur la prévalence d'anticorps anti-AAV9 dans la population générale montrent des taux faibles d'exposition antérieure à l'AAV9 dans la population pédiatrique. Une recherche d'anticorps anti-AAV9 doit être réalisée avant la perfusion d'onasemnogene abeparvovec. Une nouvelle analyse peut être effectuée si le titre d'anticorps anti-AAV9 rapporté est supérieur à 1:50. On ne sait pas encore si ou dans quelles conditions l'onasemnogene abeparvovec peut être administré de façon sûre et efficace en cas de titres d'anticorps anti-AAV9 supérieurs à 1:50 (voir rubriques Posologie et mode d'administration et Propriétés pharmacodynamiques).
SMA de forme avancée
Étant donné que la SMA entraîne une atteinte progressive et non réversible des motoneurones moteurs, le bénéfice de l'onasemnogene abeparvovec chez les patients symptomatiques dépend du degré de la maladie au moment du traitement ; plus le traitement est précoce et plus le bénéfice potentiel est significatif. Même si les patients atteints de SMA symptomatique de forme avancée n'atteindront pas les mêmes étapes de développement de la motricité globale que leurs pairs en bonne santé, ils peuvent tirer un bénéfice clinique de la thérapie génique, en fonction du stade de la maladie au moment du traitement (voir rubrique Propriétés pharmacodynamiques).
Le médecin traitant doit prendre en compte le fait que le bénéfice est très réduit chez les patients présentant une faiblesse musculaire sévère et une insuffisance respiratoire, chez les patients sous ventilation permanente et chez les patients qui ne sont pas en mesure de déglutir.
Le rapport bénéfice/risque de l'onasemnogene abeparvovec chez les patients atteints de SMA de forme avancée, maintenus en vie par la ventilation permanente et présentant une absence de croissance, n'est pas établi.
Immunogénicité
Une réponse immunitaire à la capside du vecteur viral adéno-associé de sérotype 9 (AAV9) se produira après l'administration d'onasemnogene abeparvovec, incluant la formation d'anticorps contre la capside de l'AAV9 malgré le traitement immunomodulateur recommandé à la rubrique Posologie et mode d'administration et une réponse immunitaire à médiation cellulaire.
Une réponse immunitaire systémique, y compris une hépatotoxicité d'origine immunitaire, a été rapportée dans le programme clinique de l'onasemnogene abeparvovec et elle peut nécessiter un ajustement du traitement immunomodulateur, comprenant une durée plus longue ou une augmentation de la dose. Voir la rubrique Posologie et mode d'administration pour les recommandations concernant le traitement immunomodulateur et les sous-rubriques Atteinte hépatique et Traitement immunomodulateur ci-dessous pour des informations détaillées.
Atteinte hépatique
· L'administration du vecteur AAV peut entraîner des augmentations des transaminases, qui peuvent être graves.
· Des cas d'atteinte hépatique grave sont survenus (voir rubrique Effets indésirables).
· Le risque d'atteinte hépatique peut être majoré chez les patients présentant une insuffisance hépatique préexistante ou une hépatite virale aiguë.
· Avant la perfusion, la fonction hépatique doit être évaluée chez tous les patients par un examen clinique et des bilans biologiques (par exemple, dosage des transaminases hépatiques ASAT et ALAT et de la bilirubine totale (voir rubrique Posologie et mode d'administration)).
· Pour atténuer les augmentations potentielles des transaminases, une corticothérapie systémique doit être administrée chez tous les patients avant et après la perfusion d'onasemnogene abeparvovec (voir rubrique Posologie et mode d'administration).
· La fonction hépatique doit être surveillée pendant au moins 3 mois après la perfusion.
· Les risques et les bénéfices de l'administration d'onasemnogene abeparvovec chez les patients présentant une insuffisance hépatique préexistante doivent être évalués attentivement par rapport aux risques de l'absence de traitement.
Les taux d'ASAT, d'ALAT et de bilirubine doivent être déterminés une fois par semaine pendant 30 jours, puis toutes les deux semaines pendant 60 jours supplémentaires après l'administration d'onasemnogene abeparvovec jusqu'à la fin de la période d'arrêt progressif de la corticothérapie, ou plus longtemps si nécessaire. L'arrêt progressif de la prednisolone ne doit être envisagé que lorsque les taux d'ASAT et d'ALAT sont inférieurs à 2 x LSN.
Thrombopénie
Des diminutions transitoires du taux de plaquettes, dont certaines répondaient aux critères de thrombopénie, ont été rapportées dans les études cliniques avec l'onasemnogene abeparvovec. Dans la majorité des cas, le nadir plaquettaire était observé au cours de la première semaine suivant la perfusion d'onasemnogene abeparvovec. Une numération plaquettaire doit être réalisée avant la perfusion d'onasemnogene abeparvovec et le taux de plaquettes doit être contrôlé à intervalles réguliers par la suite, une fois par semaine pendant le premier mois, puis toutes les deux semaines pendant le deuxième et le troisième mois, jusqu'à ce qu'il soit revenu à la valeur avant traitement.
Élévation de la troponine I
Des augmentations du taux de troponine I cardiaque ont été rapportées après la perfusion d'onasemnogene abeparvovec. Les taux élevés de troponine I observés chez certains patients peuvent indiquer une éventuelle atteinte du tissu myocardique. Sur la base de ces observations et de la cardiotoxicité constatée chez la souris, le taux de troponine I doit être déterminé avant la perfusion d'onasemnogene abeparvovec et surveillé pendant au moins trois mois après la perfusion d'onasemnogene abeparvovec ou jusqu'à ce qu'il soit revenu dans les valeurs normales pour les patients atteints de SMA. La consultation d'un cardiologue doit être envisagée si nécessaire.
Traitement immunomodulateur
Le traitement immunomodulateur ne doit pas être instauré chez les patients présentant des infections actives, qu'il s'agisse d'infections aiguës (telles qu'infections respiratoires aiguës ou hépatite aiguë) ou d'infections chroniques non contrôlées (telles qu'hépatite B chronique évolutive) (voir rubriques Posologie et mode d'administration et Mises en garde et précautions d'emploi).
Le traitement immunomodulateur (voir rubrique Posologie et mode d'administration) peut également diminuer la réponse immunitaire aux infections (respiratoires) concurrentes, ce qui peut entraîner une évolution clinique plus sévère de l'infection concurrente. Des précautions supplémentaires en ce qui concerne le moment d'administration d'onasemnogene abeparvovec sont recommandées en cas d'infection (virale) en phase prodromique ou en cours de résolution. Une vigilance intensifiée pour le diagnostic et la prise en charge active d'une infection respiratoire (virale) est recommandée. Les traitements prophylactiques saisonniers contre les infections par le virus respiratoire syncytial (VRS) sont recommandés et doivent être à jour. Si possible, le calendrier de vaccinations du patient doit être adapté par rapport à l'administration concomitante de corticoïdes avant et après la perfusion d'onasemnogene abeparvovec (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Le médecin traitant doit être conscient de la possibilité d'une insuffisance surrénalienne liée à la durée plus longue de la corticothérapie, ce qui peut influer sur le schéma du traitement immunomodulateur proposé.
Excrétion
Une excrétion temporaire de l'onasemnogene abeparvovec se produit, essentiellement par l'intermédiaire des déchets corporels. Les familles des patients et les aidants doivent recevoir les instructions suivantes sur la manipulation correcte des selles du patient :
· Une hygiène des mains correcte est nécessaire en cas de contact direct avec les déchets corporels du patient pendant au moins un mois après le traitement par l'onasemnogene abeparvovec.
· Les couches jetables peuvent être placées dans des doubles sacs en plastique fermés et jetées avec les ordures ménagères.
Ce médicament contient du sodium
Ce médicament contient 4,6 mg de sodium par mL, ce qui équivaut à 0,23 % de l'apport alimentaire quotidien maximal recommandé par l'OMS de 2 g de sodium par adulte. Chaque flacon de 5,5 mL contient 25,3 mg de sodium et chaque flacon de 8,3 mL contient 38,2 mg de sodium.
Résumé du profil de sécurité
Les effets indésirables les plus fréquemment rapportés après l'administration étaient uneaugmentation transitoire des transaminases hépatiques (12,4 %) et des vomissements (8,2 %), voir rubrique Mises en garde et précautions d'emploi.
Liste tabulée des effets indésirables
Les effets indésirables identifiés chez tous les patients traités par l'onasemnogene abeparvovec administré en perfusion intraveineuse et ayant une relation causale avec le traitement sont présentés dans le tableau 3. Les effets indésirables sont présentés par classe de système d'organes MedDRA et fréquence. Les catégories de fréquence sont définies selon la convention suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Tableau 3 Liste tabulée des effets indésirables de l'onasemnogene abeparvovec
| Effets indésirables présentés par SOC/PT MedDRA et fréquence | |
| Affections hématologiques et du système lymphatique | |
| Fréquent | Thrombopénie |
| Affections gastro-intestinales | |
| Fréquent | Vomissements |
| Troubles généraux et anomalies au site d'administration | |
| Fréquent | Pyrexie |
| Investigations | |
| Très fréquent | Augmentation des transaminases |
| Fréquent | Aspartate aminotransférase augmentée, alanine aminotransférase augmentée, troponine-I augmentée |
Description de certains effets indésirables
Affections hépatobiliaires
Des augmentations des transaminases supérieures à 2 x LSN ont été rapportées chez près de 12 % des patients traités à la dose recommandée et elles ont été jugées comme étant liées au médicament expérimental. Deux patients avaient des taux d'ASAT et d'ALAT > 20 x LSN (un de ces patients présentait une infection virale). Ces patients étaient cliniquement asymptomatiques, ne présentaient pas d'ictère ni d'augmentation cliniquement significative de la bilirubine et ne répondaient pas aux critères de la loi de Hy. Les augmentations des transaminases sériques se sont résolues avec un traitement par prednisolone (voir rubriques Posologie et mode d'administration et Mises en garde et précautions d'emploi) et les patients ont récupéré sans séquelles cliniques.
En dehors des études cliniques, un cas d'atteinte hépatique aiguë grave a été rapporté avec l'onasemnogene abeparvovec ; le patient continuait à recevoir le traitement par le nusinersen et présentait des élévations de l'ASAT et de l'ALAT > 3 x LSN avant le traitement par l'onasemnogene abeparvovec. Il s'est rétabli grâce à une corticothérapie supplémentaire.
Thrombopénie transitoire
Des diminutions transitoires du taux de plaquettes moyens par rapport à la valeur initiale (4,1 %) ont été observées à plusieurs temps de mesure après la perfusion ; elles se sont généralement résolues en deux semaines. Les diminutions du taux de plaquettes étaient plus importantes au cours de la première semaine de traitement. Aucun patient n'a présenté de symptômes cliniques associés à la diminution des plaquettes (voir rubrique Mises en garde et précautions d'emploi).
Augmentation du taux de troponine I
Des augmentations du taux de troponine I cardiaque (3,1 %) allant jusqu'à 0,2 µg/L ont été observées après la perfusion d'onasemnogene abeparvovec. Dans le programme d'études cliniques, il n'a pas été observé d'anomalies cardiaques cliniquement manifestes après l'administration d'onasemnogene abeparvovec (voir rubrique Mises en garde et précautions d'emploi).
Immunogénicité
Les titres d'anticorps anti-AAV9 avant et après l'administration du produit de thérapie génique étaient mesurés dans les études cliniques (voir rubrique Mises en garde et précautions d'emploi). Tous les patients ayant reçu l'onasemnogene abeparvovec avaient des titres d'anticorps anti-AAV9 inférieurs ou égaux à 1:50 avant le traitement. Chez tous les patients, des augmentations du titre moyen d'anticorps anti-AAV9 ont été observées à tous les temps sauf un de dosage des anticorps dirigés contre le peptide de l'AAV9, ce qui reflète la réponse normale à un antigène viral du non-soi. Certains patients avaient des titres d'anticorps anti-AAV9 supérieurs au seuil de quantification ; cependant, la majorité de ces patients n'a pas présenté d'effets indésirables potentiellement cliniquement significatifs. Par conséquent, il n'a pas été établi de relation entre des titres élevés d'anticorps anti-AAV9 et le risque d'effets indésirables ou les paramètres d'efficacité.
Dans l'étude clinique AVXS-101-CL-101, une recherche d'anticorps anti-AAV9 a été effectuée chez 16 patients : 13 patients avaient un titre inférieur à 1:50 et ont été inclus dans l'étude. Trois patients avaient un titre supérieur à 1:50 ; un nouveau dosage a été réalisé après l'arrêt de l'allaitement, les titres étaient inférieurs à 1:50 et les deux patients ont été inclus dans l'étude. Il n'existe pas de données indiquant si l'allaitement doit ou non être limité chez les femmes susceptibles d'être séropositives pour les anticorps anti-AAV9. Tous les patients avaient un titre d'anticorps anti-AAV9 ≤ 1:50 avant le traitement par l'onasemnogene abeparvovec et ont présenté ensuite une augmentation attendue du titre d'anticorps à au moins 1:102 400 et allant jusqu'à plus de 1:819 200.
La détection de formation d'anticorps dépend fortement de la sensibilité et de la spécificité de la méthode de dosage. De plus, plusieurs facteurs, incluant la méthode analytique, la manipulation de l'échantillon, le moment du prélèvement de l'échantillon, les médicaments concomitants et la maladie sous-jacente, peuvent influer sur l'incidence de positivité pour les anticorps (y compris les anticorps neutralisants) observée dans un dosage.
Aucun patient traité par l'onasemnogene abeparvovec n'a présenté de réponse immunitaire contre le transgène.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté au moyen de la fiche correspondante (voir Annexe D) du protocole d'utilisation thérapeutique.
SURVEILLANCE
avant traitement :
- recherche
d'anticorps anti AAV9 à l'aide d'un dosage validé de façon appropriée, une nouvelle
analyse peut être effectuée si le titre d'anticorps anti-AAV9 rapporté est
supérieur à 1:50.
- fonction hépatique : alanine aminotransférase (ALAT), aspartate aminotransférase (ASAT) et bilirubine totale,
- numération plaquettaire,
- dosage de la troponine I.
- avant chaque administration : rechercher de symptômes de maladie infectieuse active de toute nature.
SURVEILLANCE pendant le traitement :
- fonction hépatique : taux d'ASAT, d'ALAT et de bilirubine à déterminer une fois par semaine pendant 30 jours, puis toutes les deux semaines pendant 60 jours supplémentaires après l'administration d'onasemnogene abeparvovec jusqu'à la fin de la période d'arrêt progressif de la corticothérapie, ou plus longtemps si nécessaire,
- taux de plaquettes à contrôler à intervalles réguliers, une fois par semaine pendant le premier mois, puis toutes les deux semaines pendant le deuxième et le troisième mois, jusqu'à ce qu'il soit revenu à la valeur avant traitement,
- taux de troponine I à surveiller pendant au moins trois mois après la perfusion d'onasemnogene abeparvovec ou jusqu'à ce qu'il soit revenu dans les valeurs normales. La consultation d'un cardiologue doit être envisagée si nécessaire.
PROPHYLAXIE :
Une immunomodulation par corticoïdes est recommandée pour diminuer la réponse immunitaire. Si possible, le calendrier de vaccinations du patient doit être adapté par rapport à l'administration concomitante de corticoïdes avant et après la perfusion d'onasemnogene abeparvovec. Une prophylaxie saisonnière contre les infections par le VRS est recommandée. (Les vaccins à virus vivant tels que le vaccin ROR et le vaccin contre la varicelle ne doivent pas être administrés chez les patients recevant une dose de corticoïde immunosuppressive (c'est-à-dire ≥ 2 semaines d'administration quotidienne de 20 mg ou de 2 mg/kg de poids corporel de prednisone ou équivalent).
INFORMATIONS à communiquer aux familles des patients et aux aidants concernant la manipulation correcte des selles du patient :
- Une hygiène des mains correcte est nécessaire en cas de contact direct avec les déchets corporels du patient pendant au moins un mois après le traitement par l'onasemnogene abeparvovec.
- Les couches jetables peuvent être placées dans des doubles sacs en plastique fermés et jetées avec les ordures ménagères.
CONSULTER un médecin en urgence si l'enfant présente :
- ecchymoses ou saignements durant plus longtemps que d'habitude après une blessure,
- peau de couleur gris pâle ou bleue, difficultés respiratoires (par exemple, respiration rapide,
essoufflement), gonflement des membres ou de l'abdomen.
INFORMER immédiatement le médecin ou l'infirmière de l'enfant si l'enfant présente l'un de ces symptômes :
- toux, respiration sifflante, éternuements, écoulement nasal, mal de gorge ou fièvre.
- augmentations des enzymes hépatiques (transaminases) montrées par les analyses de sang.
- vomissements ;
- fièvre.
Il n'y a pas de données chez la femme concernant l'utilisation de l'onasemnogene abeparvovec pendant la grossesse ou l'allaitement.
Il n'y a pas eu d'études de fertilité ni d'étude de reproduction conduites avec l'onasemnogene abeparvovec chez l'animal.
Aucune étude d'interaction n'a été réalisée.
L'expérience de l'utilisation de l'onasemnogene abeparvovec chez des patients recevant un traitement hépatotoxique ou utilisant des substances hépatotoxiques est limitée. La sécurité de l'onasemnogene abeparvovec chez ces patients n'a pas été établie.
L'expérience de l'utilisation concomitante d'agents ciblés pour le traitement de la SMA 5q est limitée.
Vaccinations
Si possible, le calendrier de vaccinations du patient doit être adapté par rapport à l'administration concomitante de corticoïdes avant et après la perfusion d'onasemnogene abeparvovec (voir rubriques Posologie et mode d'administration et Mises en garde et précautions d'emploi). Une prophylaxie saisonnière contre les infections par le VRS est recommandée (voir rubrique Mises en garde et précautions d'emploi). Les vaccins à virus vivant tels que le vaccin ROR et le vaccin contre la varicelle ne doivent pas être administrés chez les patients recevant une dose de corticoïde immunosuppressive (c'est-à-dire ≥ 2 semaines d'administration quotidienne de 20 mg ou de 2 mg/kg de poids corporel de prednisone ou équivalent).
Le traitement doit être instauré et administré dans un milieu hospitalier et supervisé par un médecin expérimenté dans la prise en charge des patients atteints de SMA.
Avant l'administration d'onasemnogene abeparvovec, un bilan biologique initial incluant ce qui suit doit être réalisé :
- recherche d'anticorps anti AAV9 à l'aide d'un dosage validé de façon appropriée ;
- fonction hépatique : alanine aminotransférase (ALAT), aspartate aminotransférase (ASAT) et bilirubine totale ;
- numération plaquettaire ; et
- dosage de la troponine I.
La nécessité d'une surveillance étroite de la fonction hépatique et des taux de plaquettes et de troponine I après l'administration et la nécessité d'une corticothérapie doivent être prises en compte pour programmer le moment du traitement par l'onasemnogene abeparvovec (voir rubrique Mises en garde et précautions d'emploi).
En cas d'infections actives aiguës ou chroniques non contrôlées, le traitement doit être différé jusqu'à ce que l'infection se soit résolue ou soit contrôlée (voir sous-rubriques Posologie et mode d'administration et Mises en garde et précautions d'emploi Traitement immunomodulateur).
Posologie
Pour perfusion intraveineuse unique exclusivement.
Les patients doivent recevoir une dose nominale de 1,1 x 1014 vg/kg d'onasemnogene abeparvovec. Le volume total est déterminé en fonction du poids du patient.
Le tableau 1 présente la dose recommandée chez les patients pesant de 2,6 kg à 21,0 kg.
Tableau 1 Dose recommandée en fonction du poids du patient
| Intervalle de poids (kg) | Dose (vg) | Volume total de la dosea (mL) |
| 2,6 à 3,0 | 3,3 x 1014 | 16,5 |
| 3,1 à 3,5 | 3,9 x 1014 | 19,3 |
| 3,6 à 4,0 | 4,4 x 1014 | 22,0 |
| 4,1 à 4,5 | 5,0 x 1014 | 24,8 |
| 4,6 à 5,0 | 5,5 x 1014 | 27,5 |
| 5,1 à 5,5 | 6,1 x 1014 | 30,3 |
| 5,6 à 6,0 | 6,6 x 1014 | 33,0 |
| 6,1 à 6,5 | 7,2 x 1014 | 35,8 |
| 6,6 à 7,0 | 7,7 x 1014 | 38,5 |
| 7,1 à 7,5 | 8,3 x 1014 | 41,3 |
| 7,6 à 8,0 | 8,8 x 1014 | 44,0 |
| 8,1 à 8,5 | 9,4 x 1014 | 46,8 |
| 8,6 à 9,0 | 9,9 x 1014 | 49,5 |
| 9,1 à 9,5 | 1,05 x 1015 | 52,3 |
| 9,6 à 10,0 | 1,1 x 1015 | 55,0 |
| 10,1 à 10,5 | 1,2 x 1015 | 57,8 |
| 10,6 à 11,0 | 1,21 x 1015 | 60,5 |
| 11,1 à 11,5 | 1,27 x 1015 | 63,3 |
| 11,6 à 12,0 | 1,32 x 1015 | 66,0 |
| 12,1 à 12,5 | 1,36 x 1015 | 68,8 |
| 12,6 à 13,0 | 1,44 x 1015 | 71,5 |
| 13,1 à 13,5 | 1,49 x 1015 | 74,3 |
| 13,6 à 14,0 | 1,54 x 1015 | 77,0 |
| 14,1 à 14,5 | 1,59 x 1015 | 79,8 |
| 14,6 à 15,0 | 1,65 x 1015 | 82,5 |
| 15,1 à 15,5 | 1,71 x 1015 | 85,3 |
| 15,6 à 16,0 | 1,76 x 1015 | 88,0 |
| 16,1 à 16,5 | 1,82 x 1015 | 90,8 |
| 16,6 à 17,0 | 1,87 x 1015 | 93,5 |
| 17,1 à 17,5 | 1,93 x 1015 | 96,3 |
| 17,6 à 18,0 | 1,98 x 1015 | 99,0 |
| 18,1 à 18,5 | 2,04 x 1015 | 101,8 |
| 18,6 à 19,0 | 2,09 x 1015 | 104,5 |
| 19,1 à 19,5 | 2,15 x 1015 | 107,3 |
| 19,6 à 20,0 | 2,20 x 1015 | 110,0 |
| 20,1 à 20,5 | 2,26 x 1015 | 112,8 |
| 20,6 à 21,0 | 2,31 x 1015 | 115,5 |
a REMARQUE : le nombre de flacons par kit et le nombre de kits nécessaires dépendent du poids. Le volume de la dose est calculé en utilisant la limite supérieure de la fourchette de poids.
Traitement immunomodulateur
Une réponse immunitaire à la capside du vecteur viral adéno-associé de sérotype 9 (AAV9) se produira après l'administration d'onasemnogene abeparvovec (voir rubrique Mises en garde et précautions d'emploi). Des augmentations des taux de transaminases hépatiques, de troponine I ou une diminution du taux de plaquettes peuvent être induites (voir rubriques Mises en garde et précautions d'emploi et Effets indésirables). Une immunomodulation par corticoïdes est recommandée pour diminuer la réponse immunitaire. Si possible, le calendrier de vaccinations du patient doit être adapté par rapport à l'administration concomitante de corticoïdes avant et après la perfusion d'onasemnogene abeparvovec (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Avant le début du traitement immunomodulateur et avant l'administration d'onasemnogene abeparvovec, le patient doit être évalué pour rechercher des symptômes de maladie infectieuse active de toute nature.
Il est recommandé d'instaurer un traitement immunomodulateur débutant 24 heures avant la perfusion d'onasemnogene abeparvovec, conformément au calendrier ci-dessous (tableau 2). Les déviations par rapport à ces recommandations sont à l'appréciation du médecin traitant (voir rubrique Mises en garde et précautions d'emploi).
Tableau 2 Traitement immunomodulateur avant et après la perfusion
| Avant la perfusion | 24 heures avant la perfusion d'onasemnogene abeparvovec | Prednisolone 1 mg/kg/jour (ou équivalent) par voie orale |
| Après la perfusion | 30 jours (incluant le jour d'administration de l'onasemnogene abeparvovec) | Prednisolone 1 mg/kg/jour (ou équivalent) par voie orale |
| 28 jours suivants : Chez les patients dont les résultats ne montrent rien de significatif (examen clinique normal, taux de bilirubine totale normale, et chez les patients dont les taux d'ALAT et d'ASAT sont tous deux inférieurs à 2 x la limite supérieure de la normale [LSN]) à la fin de la période de 30 jours ; ou | Arrêt progressif de la prednisolone orale (ou équivalent), par exemple 2 semaines à 0,5 mg/kg/jour puis 2 semaines à 0,25 mg/kg/jour | |
| Chez les patients présentant des anomalies de la fonction hépatique à la fin de la période de 30 jours : poursuite de la corticothérapie jusqu'à ce que les taux d'ALAT et d'ASAT soient inférieurs à 2 x LSN et que tous les autres paramètres soient normalisés, avec ensuite un arrêt progressif sur 28 jours. | Corticoïdes systémiques (dose équivalente à 1 mg/kg/jour de prednisolone orale) | |
| Les taux de transaminases hépatiques doivent être surveillés pendant au moins 3 mois après la perfusion d'onasemnogene abeparvovec (voir rubrique Mises en garde et précautions d'emploi). | ||
Il convient de consulter un ou plusieurs spécialiste(s) si le patient ne présente pas de réponse adéquate à l'équivalent d'1 mg/kg par jour de prednisolone orale.
Si le médecin utilise un autre corticoïde à la place de la prednisolone, les mêmes considérations s'appliquent et la même stratégie pour la diminution progressive de la dose après 30 jours doit être utilisée le cas échéant.
Populations particulières Insuffisance rénale
La sécurité et l'efficacité de l'onasemnogene abeparvovec chez les patients présentant une insuffisance rénale n'ont pas été établies et le traitement par l'onasemnogene abeparvovec doit être envisagé avec précaution. Un ajustement de la dose ne doit pas être envisagé.
Insuffisance hépatique
L'onasemnogene abeparvovec n'a pas été étudié chez les patients présentant une insuffisance hépatique. L'onasemnogene abeparvovec ne doit pas être administré, sauf si l'hyperbilirubinémie est due à un ictère néonatal. Le traitement par l'onasemnogene abeparvovec doit être envisagé avec précaution chez les patients atteints d'insuffisance hépatique (voir rubriques Mises en garde et précautions d'emploi et Effets indésirables). Un ajustement de la dose ne doit pas être envisagé.
Génotype 0SMN1/1SMN2
Aucun ajustement de la dose ne doit être envisagé chez les patients porteurs d'une mutation bi-allélique du gène SMN1 et d'une seule copie du gène SMN2 (voir rubrique Propriétés pharmacodynamiques).
Anticorps antiAAV9
Aucun ajustement de la dose ne doit être envisagé chez les patients ayant un titre d'anticorps anti-AAV9 supérieur à 1:50 avant le traitement (voir rubrique Mises en garde et précautions d'emploi).
Population pédiatrique
La sécurité et l'efficacité de l'onasemnogene abeparvovec chez les nouveau-nés prématurés avant que l'âge gestationnel à terme soit atteint n'ont pas été établies. Aucune donnée n'est disponible. L'administration d'onasemnogene abeparvovec doit être envisagée avec prudence car la corticothérapie concomitante peut avoir des effets délétères sur le développement neurologique.
L'expérience chez les patients âgés de 2 ans et plus ou pesant plus de 13,5 kg est limitée. La sécurité et l'efficacité de l'onasemnogene abeparvovec chez ces patients n'ont pas été établies. Les données actuellement disponibles sont décrites à la rubrique Propriétés pharmacodynamiques. Un ajustement de la dose ne doit pas être envisagé (voir tableau 1).
Mode d'administration
Voie intraveineuse.
L'onasemnogene abeparvovec est administré en perfusion intraveineuse unique. Il doit être administré à l'aide d'un pousse-seringue en perfusion intraveineuse unique lente d'environ 60 minutes. Il ne doit pas être administré en injection intraveineuse rapide ou en bolus.
La pose d'un second cathéter (« de secours ») est recommandée en cas d'obstruction du cathéter principal. À la fin de la perfusion, la tubulure doit être rincée avec une solution de chlorure de sodium.
Pour les instructions concernant la dilution du médicament avant administration, voir la rubrique Instructions pour l'utilisation, la manipulation et l'élimination.
Précautions à prendre avant la manipulation ou l'administration du médicament
Ce médicament contient un organisme génétiquement modifié. Un équipement de protection individuelle (incluant blouse, lunettes de sécurité et gants) doit être porté pendant la préparation ou l'administration de l'onasemnogene abeparvovec (voir rubrique Instructions pour l'utilisation, la manipulation et l'élimination). Pour les instructions concernant la préparation, la manipulation, les moyens d'éviter une exposition accidentelle et l'élimination du médicament, y compris la manipulation correcte des déchets corporels, voir la rubrique Instructions pour l'utilisation, la manipulation et l'élimination.
Durée de conservation :
1 an
Après décongélation
Après avoir été décongelé, le médicament ne doit pas être recongelé et peut être conservé au réfrigérateur à une température comprise entre 2 °C et 8 °C pendant 14 jours dans l'emballage d'origine.
Une fois le volume de la dose prélevé dans la seringue, le médicament doit être administré dans les 8 heures. Si le médicament n'est pas administré dans le délai de 8 heures, la seringue contenant le vecteur doit être éliminée.
Précautions particulières de conservation :
À conserver et transporter congelé (≤ -60 °C).
À conserver au réfrigérateur (entre 2 °C et 8 °C) immédiatement après réception. À conserver dans l'emballage d'origine.
Pour les conditions de conservation du médicament après décongélation, voir la rubrique Durée de conservation.
La date de réception doit être notée sur l'emballage d'origine avant que le produit soit placé au réfrigérateur.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Il n'existe pas de données issues des études cliniques concernant un surdosage d'onasemnogene abeparvovec. Un ajustement de la dose de prednisolone, une observation clinique étroite et une surveillance des paramètres biologiques (biochimie clinique et hématologie) afin de détecter une réponse immunitaire sont recommandés (voir rubrique Mises en garde et précautions d'emploi).
Classe pharmacothérapeutique : Autres médicaments en cas de troubles du système musculosquelettique, code ATC : M09AX09.
Mécanisme d'action
L'onasemnogene abeparvovec est un produit de thérapie génique conçu pour introduire une copie fonctionnelle du gène codant pour la protéine de survie des motoneurones (SMN1) dans les cellules transduites pour traiter la cause principale monogénique de la maladie. Il est attendu qu'en fournissant une source alternative d'expression de la protéine SMN dans les motoneurones, il favorise la survie et la fonction des motoneurones transduits.
L'onasemnogene abeparvovec est un vecteur AAV recombinant non réplicatif qui utilise une capside d'un AAV9 pour délivrer un transgène SMN humain stable et totalement fonctionnel. La capacité de la capside de l'AAV9 à traverser la barrière hémato-encéphalique et à transduire les motoneurones a été démontrée. Le gène SMN1 présent dans l'onasemnogene abeparvovec est destiné à résider dans un épisome de l'ADN dans le noyau des cellules transduites et il est attendu qu'il soit exprimé de façon stable pendant une longue période dans les cellules post-mitotiques. Le virus AAV9 n'est pas connu pour être pathogène chez l'homme. Le transgène est introduit dans les cellules cibles sous forme de molécule double brin autocomplémentaire. L'expression du transgène est induite par un promoteur constitutif (amplificateur du cytomégalovirus/promoteur hybride du gène de l'actine ß de poulet), ce qui entraîne l'expression continue et maintenue de la protéine SMN. La preuve du mécanisme d'action a été étayée par les études précliniques et par les données de distribution chez l'homme.
Efficacité et sécurité clinique
Étude de phase 3 AVXS-101-CL-303 menée chez des patients atteints de SMA de type 1
L'étude AVXS-101-CL-303 (étude 303) est une étude de phase III à dose unique en ouvert, à un seul bras, de l'administration intraveineuse d'onasemnogene abeparvovec à la dose thérapeutique (1,1 x 1014 vg/kg). Vingt-deux patients atteints de SMA de type 1 et porteurs de deux copies du gène SMN2 ont été inclus. L'âge des patients lors de l'administration allait de 0,5 à 5,9 mois. Sur les 22 patients inclus, trois patients sont sortis de l'étude, dont deux patients ayant présenté un événement (décès ou mise sous ventilation permanente), soit un taux de survie sans événement de 90,9 % (IC à 95 % : 79,7 % ; 100,0%) (patients en vie sans ventilation permanente) à l'âge de 14 mois, voir figure 1.
![]() Figure
1 Délai
(jours) jusqu'au décès ou jusqu'à la mise sous ventilation permanente -
Données combinées des études de l'onasemnogene abeparvovec IV (CL-101,
CL-302, CL-303, cohorte 2 copies
Figure
1 Délai
(jours) jusqu'au décès ou jusqu'à la mise sous ventilation permanente -
Données combinées des études de l'onasemnogene abeparvovec IV (CL-101,
CL-302, CL-303, cohorte 2 copies
'étud yy tt ii ll ii bb aa bb oo rr PP ll aa vv ii vv rr uu SS
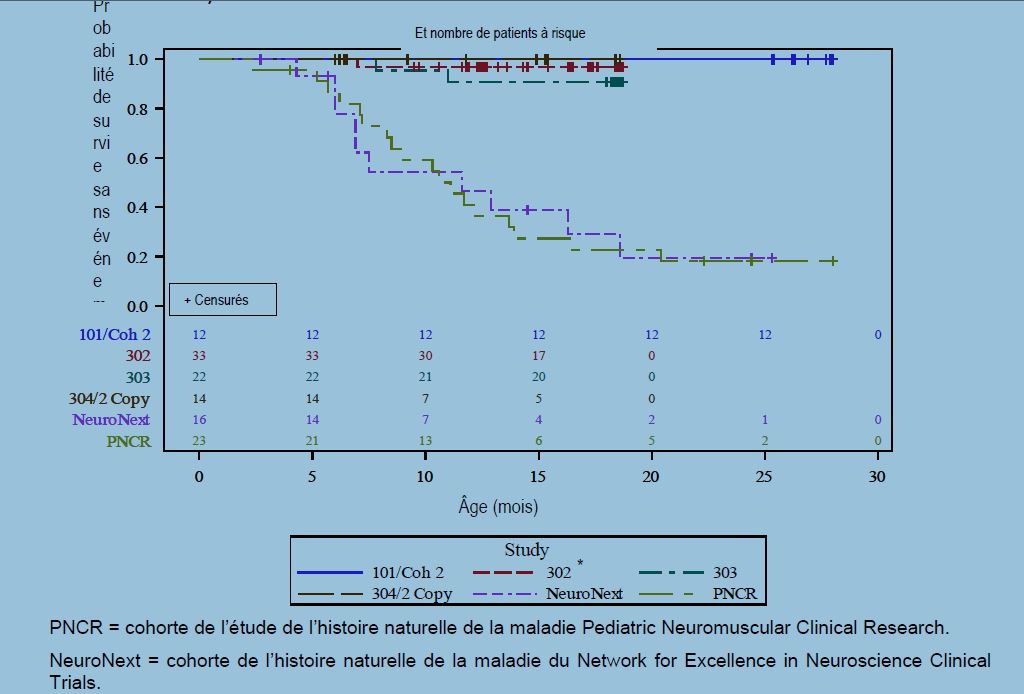
L'étude AVXS-101-CL-302 est une étude de phase III en cours, multicentrique, en ouvert, en un seul bras, à dose unique, de l'AVXS-101 (produit de thérapie génique) chez des patients atteints de SMA de type 1 avec une ou deux copies du gène SMN2 qui est similaire à l'étude AVXS-101-CL-303. L'âge moyen des patients dans l'étude au moment du gel des données le 31 décembre 2019 est de 10,62 mois (plage : 1,8 à 5,4 mois).
.
Chez les 14 patients de l'étude CL-303 qui avaient atteint l'étape de se tenir assis sans assistance pendant au moins 30 secondes, l'âge médian lors de l'atteinte de cette étape pour la première fois était de 12,5 mois (plage : 9,2 à 18,6 mois). Chez 13 patients, l'acquisition de la capacité à se tenir assis sans assistance pendant au moins 30 secondes a été confirmée lors de la visite à l'âge de 18 mois (critère co-principal, p < 0,0001). Un patient avait atteint l'étape de se tenir assis sans assistance pendant 30 secondes à l'âge de 16 mois, mais cela n'a pas été confirmé lors de la visite à l'âge de 18 mois. Le tableau 4 présente une synthèse de l'atteinte des grandes étapes du développement confirmée par vidéo chez les patients de l'étude CL-303. Trois patients (13,6 %) n'ont atteint aucune étape du développement moteur et six patients (27,2 %) avait acquis le contrôle de la tête comme étape maximale du développement moteur avant la visite d'étude finale à l'âge de 18 mois.
Tableau
4 Délai médian jusqu'à l'atteinte des grandes étapes
du développement moteur documentée par vidéo - Étude
30312
12
12
12
12
12
0 33
33
30
17
0 22
22
21
20
0 14
14
7
5
0 16
14
7
4
2
1
0 23
21
13
6
5
2
| Grande étape documentée par vidéo | Nombre de patients ayant atteint l'étape n/N (%) | Âge médian lors de l'atteinte de l'étape (mois) | Intervalle de confiance à 95 % |
| Contrôle de la tête | 17/20 (85) | 6,8 | (4,77 ; 7,17) |
| Roulade du dos sur le côté | 13/22 (59) | 11,5 | (7,77 ; 14,53) |
| Se tenir assis sans soutien pendant 30 secondes (Bayley) | 14/22 (64) | 12,5 | (10,17 ; 15,20) |
| Se tenir assis sans soutien pendant au moins 10 secondes (OMS) | 14/22 (64) | 13,9 | (11,00 ; 16,17) |
* Deux patients avaient acquis le contrôle de la tête selon l'évaluation effectuée par le clinicien lors de l'inclusion.
Un patient (4,5 %) pouvait également marcher avec une aide à l'âge de 12,9 mois. Sur la base de l'histoire naturelle de la maladie, il n'aurait pas été attendu que les patients qui répondaient aux critères d'inclusion dans l'étude deviennent capables de s'asseoir sans assistance.
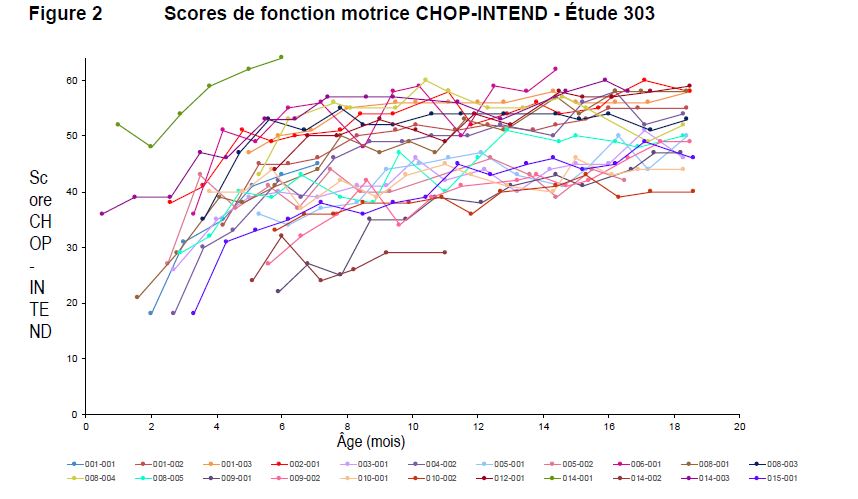
Des améliorations de la fonction motrice, mesurée par le score CHOP-INTEND (Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders), ont également été observées, voir figure 2. Vingt-et-un patients (95,5 %) avaient obtenu un score CHOP-INTEND ≥ 40, 14 patients (64 %) un score ≥ 50 et 5 patients (23 %) un score ≥ 60. Les patients atteints de SMA de type 1 non traitée n'atteignent presque jamais un score CHOP-INTEND ≥ 40. Une atteinte des grandes étapes du développement moteur a été observée chez certains patients malgré le plafonnement du score CHOP-INTEND. Il n'a pas été observé de corrélation claire entre les scores CHOP-INTEND et l'atteinte des grandes étapes du développement moteur.
Figure 2 Scores de fonction motrice CHOP-INTEND - Étude 303
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() Étude de phase I AVXS-101-CL-101 menée chez des patients atteints de SMA de type 1
Étude de phase I AVXS-101-CL-101 menée chez des patients atteints de SMA de type 1
Les résultats observés dans l'étude 303 sont corroborés par ceux de l'étude AVXS-101-CL-101 (étude de phase I menée dans la SMA de type 1, étude 101), dans laquelle l'onasemnogene abeparvovec a été administré en perfusion intraveineuse unique chez 12 patients pesant de 2,6 kg à 8,5 kg (âgés de 0,9 à 7,9 mois). À l'âge de 14 mois, tous les patients traités étaient sans événement, c'est-à-dire qu'ils survivaient sans ventilation permanente, contre 25 % des patients de la cohorte de l'étude de l'histoire naturelle de la maladie. À la fin de l'étude (24 mois après le traitement), tous les patients étaient sans événement, par rapport à moins de 8 % dans l'étude de l'histoire naturelle de la maladie ; voir figure 1.
À 24 mois de suivi après le traitement, 10 des 12 patients étaient capables de se tenir assis sans soutien pendant ≥ 10 secondes, 9 patients pouvaient se tenir assis sans soutien pendant ≥ 30 secondes et 2 patients étaient capables de se tenir debout seuls et de marcher sans assistance. Un des 12 patients n'avait pas acquis le contrôle de la tête avant l'âge de 24 mois comme étape maximale du développement moteur. Dix des 12 patients de l'étude CL-101 qui avaient reçu la dose thérapeutique proposée d'onasemnogene abeparvovec continuent à être suivis dans une étude à long terme (d'une durée allant jusqu'à 5,7 ans après le traitement) ; ils ont tous conservé les acquisitions antérieures, voire même atteint de nouvelles étapes, incluant la capacité à se tenir assis avec un soutien, à se tenir debout avec une assistance et à marcher seuls. Quatre des 10 patients ont reçu un traitement concomitant par le nusinersen à un certain moment pendant l'étude à long terme. Par conséquent, le maintien de l'efficacité et l'atteinte des étapes peuvent ne pas être imputés uniquement à l'onasemnogene abeparvovec chez tous les patients. La capacité à se tenir debout sans assistance a été acquise récemment par deux patients qui ne recevaient pas le nusinersen.
Étude de phase III AVXS-101-CL-304 menée chez des patients atteints de SMA de type 1 pré-symptomatique
L'étude CL-304 est une étude de phase III internationale en cours, à dose unique, en ouvert, à un seul bras, de l'AVXS-101 administré par voie IV chez des nouveau-nés pré-symptomatiques d'un âge allant jusqu'à 6 semaines porteurs de 2 (cohorte 1, n = 14) ou 3 (cohorte 2, n = 15) copies du gène SMN2.
Cohorte 1
Au moment de la visite de l'étude la plus récente avant le 31 décembre 2019, les patients traités porteurs de 2 copies du gène SMN2 étaient âgés de 6 mois à 18,6 mois et la durée moyenne de leur participation à l'étude était de 10,5 mois (plage : 5,1 à 18,0 mois). Tous les patients étaient en vie et sans ventilation permanente.
Huit patients avaient atteint l'étape de se tenir assis sans soutien pendant au moins 30 secondes à un âge allant de 6,4 à 11,8 mois, 7 de ces 8 enfants (87,5 %) ayant atteint l'étape de se tenir assis sans soutien avant l'âge de 9,2 mois, le 99e percentile pour l'atteinte de cette étape. Quatre patients (28,6 %) avaient atteint l'étape de marcher seuls. À la date du gel des données le 31 décembre 2019, 12 patients (85,7%) avaient obtenu un score CHOP-INTEND ≥ 60.
Cohorte 2
Au moment de la visite de l'étude la plus récente avant le 31 décembre 2019, les patients traités porteurs de 3 copies du gène SMN2 étaient âgés de 3,3 mois à 15,1 mois et la durée moyenne de leur participation à l'étude était de 8,74 mois (plage : 2 à 13,9 mois). Tous les patients étaient en vie et sans ventilation permanente. Dix des 15 patients étaient capables de se tenir assis sans soutien pendant au moins 30 secondes, 4 patients pouvaient se tenir debout sans soutien pendant au moins 3 secondes et 2 patients pouvaient monter ou descendre seuls au moins cinq marches d'escalier.
La durée de suivi est trop courte pour évaluer le développement des patients traités par l'AVXS-101 par rapport à l'histoire naturelle des patients porteurs de 3 copies du gène SMN2, chez lesquels le tableau clinique est hétérogène. Par conséquent, il n'est pas possible pour le moment de tirer de conclusions définitives sur le bénéfice dans cette population de patients.
L'onasemnogene abeparvovec n'a pas été étudié dans des études cliniques chez des patients porteurs d'une mutation bi-allélique du gène SMN1 et d'une seule copie du gène SMN2.
Des études d'excrétion du vecteur onasemnogene abeparvovec visant à évaluer la quantité de vecteur éliminée de l'organisme dans la salive, les urines et les fèces ont été réalisées.
L'onasemnogene abeparvovec était détectable dans les échantillons d'excréta après la perfusion. Il était éliminé essentiellement dans les fèces, la majeure partie étant éliminée dans les 30 jours suivant l'administration de la dose. Les concentrations d'onasemnogene abeparvovec dans les urines et la salive représentaient 0,1 % à 0,01 % de la concentration initiale dans l'organisme le jour 1 suivant la perfusion et diminuaient ensuite.
La biodistribution a été évaluée chez deux patients qui sont décédés respectivement à 5,7 mois et 1,7 mois après la perfusion d'onasemnogene abeparvovec à la dose de 1,1 x 1014 vg/kg. Dans les deux cas, les taux les plus élevés d'ADN du vecteur ont été observés dans le foie. L'ADN du vecteur a également été détecté dans la rate, le coeur, le pancréas, le ganglion inguinal, les muscles squelettiques, les nerfs périphériques, les reins, les poumons, les intestins, la moelle épinière, le cerveau et le thymus. L'immunomarquage de la protéine SMN a montré une expression généralisée de la protéine dans les motoneurones rachidiens, les neurones et les cellules gliales du cerveau et dans le coeur, le foie, les muscles squelettiques et les autres tissus analysés.
ZOLGENSMA n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Après administration par voie intraveineuse chez des souris nouveau-nées, il a été observé que le vecteur était largement distribué de façon systématique ;l'expression du transgène étant la plus élevée au niveau du coeur et le foie, avec une expression importante du transgène dans le cerveau et la moelle épinière.
Dans les études pivots de toxicologie conduites chez la souris, les principaux organes cibles de toxicités identifiés ont été le coeur et le foie.
Au niveau du coeur, des anomalies de type inflammation dose-dépendante, oedème et fibrose ont été observées dans les ventricules. Une inflammation, une thrombose, une dégénérescence/nécrose myocardique ainsi qu'une fibroplasie ont également été observées dans les oreillettes.
Au niveau hépatique, des anomalies de type hypertrophie hépatocellulaire, activation des cellules de Kupffer et nécrose hépatocellulaire disséminée ont été identifiées.
Une NOAEL n'a pu être déterminée au cours de cette étude chez la souris car les observations de type inflammation, oedème et fibrose ventriculaires et inflammation auriculaire ont été constatées à la dose la plus faible testée (1,5 x 1014 vg/kg). Cette dose est considérée comme la dose maximale tolérée et représente environ 1,4 fois la dose d'onasemnogene abeparvovec recommandée chez l'homme.
La mortalité observée chez les souris traitées à la dose de 2,4 x 1014 vg/kg a été considérée comme liée à des phénomènes de thrombose auriculaire. La cause de la mortalité chez les autres animaux n'a pas pu être établie, bien qu'un processus de dégénérescence/régénération ait été observé dans le coeur de ces animaux à l'histologie.
Il n'a pas été réalisé d'études de génotoxicité, de cancérogenèse et de toxicité de la reproduction avec l'onasemnogene abeparvovec.
Dans une étude de toxicologie menée chez de jeunes primates adultes non humains, l'administration d'une dose unique de 3 x 1013 vg (dose médiane de 1,08 x 1013 vg/kg) d'onasemnogene abeparvovec par voie intrathécale (en position de Trendelenburg), sans administration de corticoïdes, a entraîné une infiltration inflammatoire minime à marquée de cellules mononuclées (lymphocytes principalement) dans certains ganglions de la racine dorsale à tous les niveaux de la moelle épinière examinés, avec une satellitose péri-neuronale, une nécrose neuronale ou une perte neuronale complète avec minéralisation dans de rares cas. La pertinence clinique de ces observations n'est pas connue.
Ce médicament contient des cellules génétiquement modifiées. Il faut suivre les recommandations locales en matière de biosécurité qui sont applicables à la manipulation des médicaments contenant des organismes génétiquement modifiés.
· ZOLGENSMA doit être préparé de manière aseptique en Enceinte Sécurité Biologie de catégorie II dans des conditions stériles. Les surfaces de travail et le matériel ayant potentiellement été en contact avec ZOLGENSMA doivent être décontaminés avec un désinfectant approprié.
Réception et décongélation des flacons de ZOLGENSMA
· Les flacons seront transportés congelés (≤ -60 °C). À réception, les flacons doivent être mis immédiatement au réfrigérateur à une température comprise entre 2 °C et 8 °C, dans la boîte d'origine. Le traitement par l'onasemnogene abeparvovec doit être administré dans les 14 jours suivant la réception des flacons.
· Les flacons doivent être décongelés avant utilisation. N'utiliser ZOLGENSMA que s'il est décongelé.
· Pour les boîtes contenant jusqu'à 9 flacons, le produit sera décongelé après environ 12 heures au réfrigérateur. Pour les boîtes contenant jusqu'à 14 flacons, le produit sera décongelé après environ 16 heures au réfrigérateur. Sinon, et pour une utilisation immédiate, la décongélation peut être effectuée à température ambiante.
· Pour les boîtes contenant jusqu'à 9 flacons, le produit sera décongelé après environ 4 heures à température ambiante (entre 20 °C et 25 °C°). Pour les boîtes contenant jusqu'à 14 flacons, le produit sera décongelé après environ 6 heures à température ambiante (entre 20 °C et 25 °C°).
· Avant de prélever le volume de la dose dans la seringue, faire tourner doucement le produit décongélé. Ne PAS agiter.
· N'utilisez pas ce médicament si vous remarquez la présence de particules ou une coloration anormale après la décongélation du produit et avant son administration.
· Après avoir été décongelé, le médicament ne doit pas être recongelé.
· Après décongélation, ZOLGENSMA doit être administré dès que possible. Une fois le volume de la dose prélevé dans la seringue, la perfusion doit être administrée dans les 8 heures. Jeter la seringue contenant le vecteur si la perfusion n'a pas été administrée dans un délai de 8 heures.
Administration de ZOLGENSMA au patient
· Pour administrer ZOLGENSMA, prélevez la totalité du volume de la dose dans la seringue. Retirez l'air présent dans la seringue avant la perfusion intraveineuse par cathéter veineux inséré dans un membre périphérique du patient. L'insertion d'un cathéter secondaire (« back-up ») est recommandée en cas de blocage du cathéter primaire.
· ZOLGENSMA est administré par voie intraveineuse en 1 heure environ. Il doit être administré en perfusion intraveineuse exclusivement. Il ne doit pas être administré en bolus intraveineux. Après la fin de la perfusion, la voie doit être rincée avec une solution saline.
Médicament réservé à l’usage hospitalier
Prescription réservée aux spécialistes en neurologie et en pédiatrie.
Médicament nécessitant une surveillance particulière pendant le traitement.
Solution pour perfusion.
Après décongélation, le produit est une solution limpide à légèrement opaque incolore à blanchâtre.
ZOLGENSMA est présenté dans un flacon (flacon en polymère Crystal Zenith de 10 mL) muni d'un bouchon (caoutchouc chlorobutyle de 20 mm) et d'une capsule (de type flip-off en aluminium) avec un opercule de couleur (en plastique), en deux volumes de remplissage différents, 5,5 mL ou 8,3 mL.
La dose d'onasemnogene abeparvovec et le nombre exact de flacons nécessaire pour chaque patient sont calculés en fonction du poids du patient (voir la rubrique Posologie et mode d'administration et le tableau 5 ci-dessous).
Tableau 5 Composition des boîtes/kits
| Poids du patient (kg) | Flacon de 5,5 mLa | Flacon de 8,3 mLb | Nombre total de flacons par boîte |
| 2,6 à 3,0 | 0 | 2 | 2 |
| Poids du patient (kg) | Flacon de 5,5 mLa | Flacon de 8,3 mLb | Nombre total de flacons par boîte |
| 3,1 à 3,5 | 2 | 1 | 3 |
| 3,6 à 4,0 | 1 | 2 | 3 |
| 4,1 à 4,5 | 0 | 3 | 3 |
| 4,6 à 5,0 | 2 | 2 | 4 |
| 5,1 à 5,5 | 1 | 3 | 4 |
| 5,6 à 6,0 | 0 | 4 | 4 |
| 6,1 à 6,5 | 2 | 3 | 5 |
| 6,6 à 7,0 | 1 | 4 | 5 |
| 7,1 à 7,5 | 0 | 5 | 5 |
| 7,6 à 8,0 | 2 | 4 | 6 |
| 8,1 à 8,5 | 1 | 5 | 6 |
| 8,6 à 9,0 | 0 | 6 | 6 |
| 9,1 à 9,5 | 2 | 5 | 7 |
| 9,6 à 10,0 | 1 | 6 | 7 |
| 10,1 à 10,5 | 0 | 7 | 7 |
| 10,6 à 11,0 | 2 | 6 | 8 |
| 11,1 à 11,5 | 1 | 7 | 8 |
| 11,6 à 12,0 | 0 | 8 | 8 |
| 12,1 à 12,5 | 2 | 7 | 9 |
| 12,6 à 13,0 | 1 | 8 | 9 |
| 13,1 à 13,5 | 0 | 9 | 9 |
| 13,6 à 14,0 | 2 | 8 | 10 |
| 14,1 à 14,5 | 1 | 9 | 10 |
| 14,6 à 15,0 | 0 | 10 | 10 |
| 15,1 à 15,5 | 2 | 9 | 11 |
| 15,6 à 16,0 | 1 | 10 | 11 |
| 16,1 à 16,5 | 0 | 11 | 11 |
| 16,6 à 17,0 | 2 | 10 | 12 |
| 17,1 à 17,5 | 1 | 11 | 12 |
| 17,6 à 18,0 | 0 | 12 | 12 |
| 18,1 à 18,5 | 2 | 11 | 13 |
| 18,6 à 19,0 | 1 | 12 | 13 |
| 19,1 à 19,5 | 0 | 13 | 13 |
| 19,6 à 20,0 | 2 | 12 | 14 |
| 20,1 à 20,5 | 1 | 13 | 14 |
| 20,6 à 21,0 | 0 | 14 | 14 |
a La concentration nominale est de 2 x 1013 génomes du vecteur/mL et le flacon contient un volume extractible d'au moins 5,5 mL.
b La concentration nominale est de 2 x 1013 génomes du vecteur/mL et le flacon contient un volume extractible d'au moins 8,3 mL.